A new study uses a powerful genetic tool called Mendelian randomization to examine the relationship between meal-related insulin secretion and body mass. The results suggest that gene variants that increase post-meal insulin release may modestly increase body mass. Upon close examination, the findings do not appear to support the hypothesis that insulin is the primary driver of obesity, but they may nevertheless provide a foot in the door for insulin.
Earlier this week, I received a number of messages from people who asked for my opinion of this study. Here it is.

The study
The study was published in the journal Clinical Chemistry and was conducted primarily by researchers at Harvard medical school, including David Ludwig, MD, PhD, who is a middle author (1).* Ludwig has been a vocal proponent of the carbohydrate-insulin hypothesis of obesity, which posits that the primary cause of obesity is the ability of refined carbohydrate to raise insulin levels, trapping fat inside fat cells and leading to fat gain and excess hunger.
To understand the study, we first need to understand the technique it applies, called Mendelian randomization (MR). The concept behind MR is that nature randomly assigns gene variants within a population, similar to how a researcher would randomly assign people to treatment or placebo groups in a randomized controlled trial. This randomization process ensures that the variable of interest is not systematically associated with confounding factors, allowing us to say something about cause-effect relationships. If we know how specific gene variants impact some trait of interest, in theory we have a pretty clean experiment that tells us about how that trait impacts related outcomes.
Let me give you an example. The scientific community, including myself, believes LDL cholesterol level impacts heart attack risk. LDL cholesterol levels are partially genetic, and we have identified multiple gene variants that impact LDL cholesterol level. Using this information, researchers can see if gene variants that decrease LDL cholesterol are associated with lower cardiovascular risk (and vice versa). If they consistently are, this provides evidence that LDL cholesterol level contributes to cardiovascular disease risk. As it turns out, LDL-decreasing variants are associated with lower cardiovascular risk, supporting the hypothesis (2).
In the current paper, the researchers applied this technique to the relationship between post-meal insulin secretion and body mass index (BMI), a measure of weight for height that correlates with body fatness. Using large sets of human genetic data, they asked whether people who have gene variants that cause greater post-meal insulin secretion also tend to have a higher BMI. As their measure of post-meal insulin, they used insulin measurements taken from volunteers 30 minutes after they had consumed a glucose (sugar) drink.
The study did not have a preregistered research plan, meaning that we cannot be confident that researchers did not tweak the data analysis plan partway through to achieve a desired result, whether intentionally or unintentionally. To be clear, I’m not suggesting that the authors did so, only pointing out that they did not take the opportunity to prove that they didn’t engage in this common practice.
The results
The authors used three different groups of gene variants to estimate the impact of post-meal insulin release on BMI (insulin-30 stringent, GSIS stringent, and insulin-30 relaxed), and they tested them in two large genetic data sets (GIANT and UK Biobank). The key result is below:
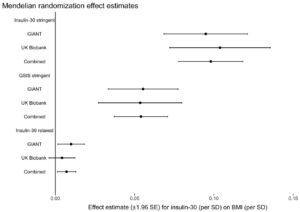
What this graph shows is that depending on the model used, one standard deviation higher post-meal insulin was associated with between about 0.01 and 0.1 standard deviation higher BMI, and this was statistically significant for most models. If I understand this correctly, roughly speaking it suggests that variation in post-meal insulin secretion explains between 1 and 10 percent of variation in BMI. About half of the larger estimate was driven by a single location of genetic variation, QPCTL/GIPR.
They then used the same method to test the reverse hypothesis– that BMI impacts meal-related insulin secretion– and did not find evidence for it, offering an argument that the previous association was not due to reverse causality.
Discussion
This study does provide evidence for a causal role of post-meal insulin release in BMI. However, before we get too carried away, we have to consider a few caveats and place the finding in the context of the rest of the scientific literature. Caveats:
- The effect size is much smaller than carbohydrate-insulin proponents generally predict. Depending on the model, post-meal insulin explains between 1 and 10 percent of BMI differences, leaving 90-99 percent unexplained by insulin. This is not consistent with insulin being a major driver of obesity, although it does get insulin’s foot in the door.
- The study measured BMI but did not directly measure body composition. We don’t know how much of the weight differences are fat, and how much are lean tissue. I would be less concerned about this if the effect size were large, because large differences in BMI are unlikely to be due to lean mass.
- The regulation of glucose, insulin, and body fatness by the brain are closely related to one another (3). Since genes that are involved in one process are often involved in the other as well, it may not be possible to separate the two functions in a genetic analysis like this, meaning that the results could be (unintentionally) misleading. Although the authors tested for this possibility, they acknowledge that due to the inherent limitations of their data set, the test may not be very informative.**
- Post-meal insulin secretion is associated with other physiological traits (possibly including leptin resistance***) that may also be influenced by the same gene variants in their models. It’s not clear that insulin itself is the variable impacting BMI, rather than another physiological variable associated with it.
When considered in the context of the rest of the literature, the current study’s finding is somewhat puzzling. For example, another recent MR study suggested that higher fasting insulin does not increase BMI, but higher BMI does increase fasting insulin (4). This result is basically the opposite of what the current study reported, but with fasting insulin swapped in for post-meal insulin. The authors of the current study suggest that the results differ because fasting insulin is simply a marker of insulin resistance, whereas post-meal insulin is a marker of insulin signaling exposure. This explanation doesn’t make much sense, for the following reasons:
- Both fasting and post-meal insulin are strong markers of insulin resistance. If a person is insulin resistant, he will generally have higher fasting and post-meal insulin levels. As a matter of fact, the insulin response to a glucose drink is commonly used as part of a formula to estimate insulin resistance (5).
- Fasting and post-meal insulin are correlated and respond to diet in concert. The amount of carbohydrate in the diet impacts both fasting and post-meal insulin. When calorie intake is matched, low-carbohydrate diets lead to lower fasting insulin in addition to lower meal-related insulin release (6).
So we are left with the puzzling fact that in the current MR study, insulin seemed to impact BMI, while in the previous study, it didn’t. I don’t know how to reconcile these two findings, and the authors’ explanation doesn’t do it for me.
Another puzzling thing about this study is the finding that BMI does not impact post-meal insulin release. The reason this is puzzling is that previous evidence shows unequivocally that it does! Check out this graph of post-glucose insulin before and after 4.5 months of moderate overfeeding (7). The closed circles are before, and the open circles are after:
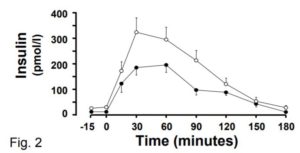
At the 30 minute time point (the one used in the current MR study), insulin secretion has nearly doubled, and this is after a modest weight increase of 2 BMI points. The fact that the current MR study did not observe this robust effect raises questions about how effective their method is at identifying cause-effect relationships in this domain.
Finally, carbohydrate-insulin advocates are using the current paper’s findings to support their larger hypothesis about the causes of obesity. However, there are still very good reasons to believe this larger hypothesis is incorrect, at least in its most prominent incarnation:
- Ludwig’s own controlled trial suggests that a low-glycemic diet causes weight loss that is virtually identical to a high-glycemic low-fat diet, despite large differences in insulin secretion (8). This is consistent with the null findings of the longest and best-controlled diet trial of glycemic index I’m aware of, conducted by one of Ludwig’s colleagues at Harvard (9).
- A 2016 study continues to be devastating to the idea that inhibiting the release of fat from fat cells increases hunger and body fatness (10). In a randomized trial, researchers gave volunteers with obesity a drug that substantially reduced fat release from fat cells, or placebo, for six months. No differences in calorie intake, macronutrient intake, metabolic rate, body weight, or body composition emerged.
- The total rate of release of fat from fat tissue in people with obesity is higher than in lean people, not lower (11). Neither insulin nor anything else is trapping fat inside the fat tissue of people with obesity, making it hard to understand how obesity could be caused by this mechanism.
- Rodents release insulin when they eat carbohydrate like humans, and their fat cells respond to insulin in the same way as humans. Yet diets that are predominantly fat tend to be more fattening in rodents than diets that are predominantly refined carbohydrate or sugar (12, 13). In fact, sugar is not very fattening at all in rodents, even at high levels. In every way that is relevant to the carbohydrate-insulin hypothesis, rodents and humans are constructed similarly, so why do rodents not respond to diet in the manner predicted by the hypothesis even under tightly controlled conditions?
- Liraglutide is a weight loss drug that mimics the hormone GLP-1. This drug was originally developed as a diabetes treatment because it stimulates insulin release, yet it was extended to obesity therapy because it substantially reduces body weight and fat (14, 15). It also improves metabolic health (16). The increase in insulin is probably not the reason why fat loss occurs, but it certainly doesn’t prevent it.
Based on these results and others, I still feel fairly confident that the carbohydrate-insulin hypothesis as formulated by Ludwig and other public advocates is not correct. But could some version of an insulin hypothesis be correct? Perhaps. The current study offers the most persuasive evidence I’ve seen so far that insulin does impact body fatness, even if only modestly. It is consistent with a model whereby multiple factors, including post-meal insulin release, determine body fatness. If the effect of insulin is small, this may explain why it’s hard to detect in other study designs such as controlled trials of low-glycemic diets. It may also imply that the effect is of little practical relevance.
Due to the caveats I discussed, I’m not ready to endorse this model yet. I’ll need to see the finding replicated independently before placing greater emphasis on it. But this study has put a foot in the door for a model of obesity that includes post-meal insulin secretion, and I’ll endorse it if future research continues to support it persuasively.
* I’m not certain what role Ludwig played in the study, but some of the language and citations in the introduction are characteristic of his writing.
** “The Egger test was not statistically significant for any of the analyses, although this test is not well-powered and may not be valid in the context of substantial heterogeneity and/or violations of the InSIDE assumption.”
*** Higher post-meal insulin release is a marker of insulin resistance, and insulin and leptin resistance tend to be associated (and likely caused by overlapping mechanisms). I would guess that people with higher post-meal insulin release are also more likely to be leptin resistant.


Yes I seem to agree on this one…if you need a test dummy I will certanly be one.
It begs the question, and I’ve no idea what the answer is. If fat should be fattening in humans as in rats, why am I and thousands like me, barrelling back fat, persistently thin, whereas when we barrelled back carbohydrate and studiously avoided fat, we were fat? My own theory is that the mixture of sugar/refined carbs and fat is not a food, but a drug to the brain and that it screws up the appestat, wherever and whatever it is.
As you’re there, were you on the panel that chose the keto diet as the worst diet of 2018?
Hi Garry,
In both humans and rats, once the fat:carbohydrate ratio reaches a certain point, weight loss occurs. For example, ketogenic diets promote weight loss in both rodents and humans (at least, in some rodent studies). High-fat diets only seem to be fattening when they include a fair amount of carbohydrate as well, and conversely, high-carbohydrate diets seem to be fattening when they include a fair amount of fat. As you said, the middle ground appears to be what is dangerous. This is not consistent with the simplistic view promoted by the CHO-insulin hypothesis, which is that carbohydrate is fattening and fat is not. That model can’t explain why very-low-fat diets are slimming and fat seems to be fattening in the middle ground of macros.
I was part of the US News and World Reports diet panel, but that doesn’t mean I agree with all the other experts’ ratings. I thought the average rating for KD was too harsh, particularly in the weight loss effectiveness rating, although I do have questions about the long-term impacts of KD. I would like to see more research to resolve those uncertainties. I plan to write a post about this at some point.
Hi Dr. Guyenet –
I would really love to see you write about the Keto diet and hunger signals in the brain.
I’m on a Keto diet, and anecdotally can still get food cravings when eating salted nuts, nut butters and low carb deserts so I opt for whole foods and emphasize protein and veggies more. Carbs aren’t the only thing that trigger over consumption for me.
I’m in a few low carb groups on Facebook and the trend has been to ditch butter coffee, eat more protein / veggies and avoid dense fatty foods (salted nuts, nut butters, fat bombs). This isn’t a universal trend, but in more and more groups people are realizing that fat can also be very palatable and lead to over consumption.
From what I’ve read in the literature – it appears that Malonyl-CoA reduces food intake (Keto increases it in the brain). Ketone esters also lower ghrelin when ingested on their own so there definitely is a reduced hunger effect that I think is independent of protein.
Something similar is probably happening to people who do intermittent fasting –
Anyway, love your work and enjoyed your book. Hope to see more frequent posting from you in the future.
I was aware of the high-fat low-fat benefit thing. I suspect that it’s due to the upper GI hormones which with the high-fat carb mixture stimulate the release of more insulin, and thus promoting obesity. This doesn’t occur to the same extent with very low fat. I think Denise Minger has a similar theory. This would explain the thin Filipinos, Chinese etc., the thin Irish in the old potato-diet etc., and conversely the thin French with much higher fat. And me. And why the SAD is utterly disastrous.
Were I you on that panel, I’d have pressed the shame button. “C’mere folks, we are dealing with a diabetic disaster and these are your recommendations? However, I’m a far more outspoken man than most and am afraid of no-one!
Your reward theory, which would be much better termed food addiction, in my view, is spot on. Carb addiction (not plain carbs, but bad carbs) is an undoubted entity and anyone who denies it is not reading up about functional MRI etc.
Are you familiar with SG’s own theory about this?
It appears you can disregard the question. 🙂 SG’s comment was not there when I initially loaded the page and I only just circled back to finish reading it – should have refreshed.
What about me and thousands like me who barrel back carbs and don’t get fat?
Having just consumed a highly rewarding lunch consisting of a grilled Tillamook smoked cheese sandwich and sweet potato cream soup, I’m glad to see that someone is still using Yudkin’s term “appestat”.
Yudkin was the most rational proponent of low carb diet IMO. He believed in counting calories AND carbs. The purpose of eating low carbs was to make low calorie meals more satiating and rewarding. Because of this better satisfaction of the appestat, Yudkin argued that a low carb dieter would also eat less fat. Yudkin vested no magical power in fat eating for weight loss.
Once weight loss is achieved there is no reason that I can see for continuing to eat a low carb diet. But there is every reason to keep counting calories to maintain weight loss.
Have you seen this:
Weight reduction through diet (1951): https://www.youtube.com/watch?v=9fxji5xkXOA
No I haven’t. Thanks. The approach used at MSU in the video is the same as what Keys developed at Minnesota and called Scientific Reducing.
These diets do not work by shifting macro ratios, but simply by reducing portion size. They preserve dietary variety.
By the late 50’s Keys had moved toward a low saturated fat diet (the precursor of his Mediterranean Diet), and Yudkin developed his low carb diet using “carbohydrate units”. In both cases the simplicity of the earlier calorie counting method was lost. In their pursuit of the optimal macros (versus simply achieving optimal weight) both men created diets that were harder to adhere to long term.
“These diets do not work by shifting macro ratios, but simply by reducing portion size. They preserve variety”.
That is the question; in the video it is claimed that to lose weight it is all about cutting calories/reducing portions, but that a diet higher in fat and protein gives better satiety, hence makes it easier both to lose weight and to keep that weight off. I could also notice different types of foods used in the experiment than what is recommended by dietitians today regardless of “macro ratios”. For example they all had 2 eggs for breakfast and three glasses of whole milk, which in my view supplies a range of valuable nutrients that could help satiety and maintain weight.
For example it has been suggested that low protein diets can lead to fatty liver. In one study it was shown that butter of beef fat in this case caused twice as much (around 30%) liver fat as olive oil and four times more than cod liver oil. However on a higher protein intake, fatty liver was prevented. Choline by itself can prevent fatty liver even on low protein intake, but saturated fats requires more choline than unsaturated. Perhaps this was a fact Ancel Keys was missing in his consideration of saturated fats. He did acknowledge the importance of leafy greens and legumes, which are good sources of folate, and could reduce need for choline, however. I would estimate in the experiment they obtained about twice as much choline as is commonly consumed by adults in the US today (2 eggs plus 3 glasses of milk supplies 400 mg alone).
It seems that some of the longest living people often ate plenty of eggs. One example is Elsa Morano who lived to be 117, she had 3 per day.
Milk may also have a slimming effect; I´ve seen anecdotes of people losing weight by following “milk only”-diets, despite a relatively high caloric intake. Not sure why, but one reason could be the higher calcium/phosphorus ratio (a low such ratio has been associated with obesity) which may affect metabolism. In the experiment they would have a higher such ratio due to all the milk and the lower intake of phosphorus rich and phytate (calcium binding) foods like nuts and whole grains. There are so many other nutrients in milk that is missing from cheese; potassium, iodine, vitamin B5 and many others. In the video it is claimed that “milk does more for the reducer than any other food”. Maybe they are right, but the project also seems to be sponsored by the dairy industry.
But my point is that there has been many dietary shifts now compared to 70 years ago; now we eat more egg whites relative to egg yolks, more cheese relative to milk and more muscle meat relative to organ meats. I assume it was hard to lose weight and hard to keep it off back then too, but my impression is that it´s even harder today, and one reason could be an improper diet, and that could involve the macrobalance (protein, fat), but also other missing nutrients. It is not like all the obese people today never tried to lose weight and think it´s fine to be have that body shape, or completely lacked willpower. Many likely lost weight at one point, but was unable to keep it off.
giuseppe the only example Yudkin gives in the book is for one day of a low calorie (ca 1000 kcal, divided into 3 meals and 3 snacks) weight loss diet. The 75g carbs would represent 30% of that, so the remaining 70% falls between protein and fat. Egg, bacon, butter, beef and grilled fish. I would assume it weights more to the fat – maybe 25% protein, 45% fat.
Except for his Carbohydrate Unit tables, Yudkin provides few details in his diet book. He leaves all the meal planning to the reader. In contrast, Keys is ALL details. His wife provided plenty of recipes and meal plans, which have complete nutrient breakdowns, and are run out for two different calorie consumption levels.
Yudkin excludes no foods in his CU tables. If you decide to consume a half pint of stout or ginger ale and you are on the 15 CU program, you have consumed half of your daily carb allocation. 75g a day of starch/sugar/alcohol isn’t much.
Just looked at Yudkin´s study published in 1960 (http://www.thelancet.com/journals/lancet/article/PIIS0140-6736(60)92019-5/abstract). In it the participants (four women, two men) were instructed to eat 50 grams of carbs and as much protein and fats as they wanted for two weeks. Caloric intake was reduced on average from about 2400 to 1400, but protein and fat intake wasn´t much changed, around 80 grams and 100 grams respectively on average.
But in % energy it would be very different. Protein intake went from 13% to 24%, fat from 41% to 63% and carbs from 46% to 13% (if I calculated correctly).
I suspect had the experiment continued longer, they would gradually have added more fat and protein as the body/liver gets adapted to it. Not sure about the long term outcome of such low carb diets. Thinking about some authors of coconut oil weight loss books that now many years after are anything but slim (or Robert Atkins). In the end I feel this endless carb vs fat war is just a distraction from the more important issue of food quality and that it´s probably better to focus on being physically active and being exposed to colder temperatures (or wearing less clothes) as a means of maintaining or reducing weight. In a study it was shown that when people moved from California to Antarctica they ended up consuming 40% more calories without losing weight: https://www.ncbi.nlm.nih.gov/pubmed/2318952).
Thanks for the Lancet link giuseppe. Yudkin was not very thorough, was sloppy with statistics, and used small study groups. But I think he was a genius in his own way, and a better communicator to the general public than Keys. This Slimming Business is illustrated with cartoonish art and quips from Ogden Nash’s doggerel poems. Yet it only contains one graph (pg 57), which has units on the y axis (food intake calories) but not the x axis (exercise). Th graph relates the function of the appestat to level of physical activity. Yudkin’s point is that at a sedentary effort level the appestat fails to regulate food intake. But what is a “sedentary”, as opposed to a “moderate” level of activity? Units are needed, not words.
so, Guiseppe, the mechanism by which choline prevents fatty liver is through more methyl donors for Phase II liver work, or is choline needed in fat metabolism? And why should folates (or vitamins B6 and 12) matter at all in methylation, when they are measured in mcg, whereas choline (and betaine) are measured in hundreds of mg? And as molecules they are quite methyl-rich, betaine having 3 per molecule.
Folate itself itsn´t a donor, rather folate coenzymes. The whole issue is too difficult for me to explain, nor fully understand (I have not much background in biochemistry). You may want to check out some of Chris Masterjohn´s articles explaining these complexities in layman´s terms. And perhaps articles like this: https://www.ncbi.nlm.nih.gov/pubmed/28460180.
So folate can spare choline, but it can´t replace it. I noticed a recent study pregnant women supplemented with 930 mg choline/d resulted in better information processing speed in the infant (than 480 mg/d). http://www.fasebj.org/content/early/2017/12/07/fj.201700692RR. Average intake among US adult women is approx 300 mg/day. But they generally obtain high amounts of folic acid as it is typically supplemented during pregnancy and many foods are now fortified with folic acid. As a result even a 2400 kcal serving of a commercial pizza supplies over 800 mcg folic acid (http://nutritiondata.self.com/facts/fast-foods-generic/9326/2), compared to just 170 mcg in a 2400 kcal human milk sample.
However, I don´t think supplements equals foods. And often we are exaggerating the presumed need for one nutrient as a result of a suboptimal intake of others. Back to the MSC experiment: 2 eggs plus 3 glasses of milk supplies “only” 60 mcg folate, but this is still many times more than what is found in a comparable (protein) amount of muscle meat, cheese or egg whites. 100 grams of misc organ meats may supply perhaps around 75 mcg folate.
An excess of sugar seems to easily cause fatty liver. But so can probably fruits. In ancient times the foie gras, fatty goose liver, was made by feeding the goose large amounts of figs, whereas today it´s more common to force feed them large amount of corn starch. But wheat supplies a fair amount of betaine and protein, so in this sense wheat would be better than sugar and maybe even fruits, for fatty liver.
I´m wondering if fatty liver could have been a relatively normal thing for humans in the stone age resulting from a temporarily high availability of honey, fruits, fatty meat (saturated) etc, which was then effectively removed during leaner times where more protein was available relative to fat and sugar (it would make an excess of protein less toxic if it was “diluted” with the liver fat).
So what Yudkin and Keys – as well as many modern researchers – could be missing is the importance of protein and choline/betaine. It may be that it´s a lack of those that would make saturated fats and sugar particularly harmful.
In the 1930´s it seems that a milk and fruit diet was sometimes used as a reducing regime (and for diabetics), see for example:
https://scholarworks.umass.edu/cgi/viewcontent.cgi?article=2549&context=theses
I suspect such a diet would work better than what many dietitians would prescribe today.
Btw, if you´re interested I made a detailed comparison between the nutrients in organ meats vs muscle meat, and milk vs cheese here: https://thepaleodiet.com/north-american-plains-indians-tall-and-robust-meat-eaters-but-not-a-milk-drinker-among-them/#comment-354419
guiseppe, when I look at Yudkin’s 75g carb daily meal plan, it strikes me that he has replaced most of the carbohydrates with protein. He took fat kcals lower than a normal diet too.
As I recall, the recent USDA dietary macrontrient in the US is 12% protein, 45% carbs and 43% fat. We are getting fat on a diet high in fat and carbs. When you look at the standard diet of the 1950’s the % fat was much lower – down in the 30% range.
Problem is that most people do not tolerate very much protein, so it´s not really practically possible to for example “replace” 300 grams of carbs with 300 grams of protein (same amount of calories). And grains are a major source of protein in most people´s diets. If this is avoided, then more protein from other sources, typically meat and dairy, needs to be added. I doubt the protein intake on Yudkin´s plan went much above 100 grams/day, certainly not above 150 grams.
What type of ratios did he recommend for weight maintenance?
I’m unaware that Yudkin counted calories and I’ve read a lot of his stuff. Counting calories is NOT the right approach, fundamentally, as it disregards satiety. I don’t count them at all and I’m 3 stone down for 4 years and my perfect weight. Magic Fairyland is when your intake is low dictated by perfect satiety. I trust my appetite so much after this period of time that some days I’m ravenous and eat to it, and other days not. My body knows what it wants. I have fat genes or whatever. All my father’s family were really fat when no-one was and his mother and one brother died of diabetes type 2. So, I have to restrict carbs.
Read Yudkin’s 1958 book This Slimming Business. Yudkin provides several pages of his 5 gram “carbohydrate units” on pages 156-158 and 183-188. These tables also contain recommended portion sizes for all foods, which put the limits on calories too. He sets CU’s at 15 for a typical slimming diet. 75g per day of simple carbs (sugars, starches and also alcohol).
Yudkin’s diet is based on counting. You have to keep a logbook for months to make it work.
Unlike many diet book authors Yudkin only supplies one sample daily diet menu, on pg 160, and shows a similar high carb low fat diet for comparison. Both diets supply about 1000 kcal. By allowing the dieter to eat bacon and eggs, beef and grilled fish it rebalances the limited calories to a much higher % protein. The dieter is left to develop their own menus using Yudkin’s CU tables.
While This Slimming Business is hard to find, Yudkin’s methodology was adopted into the ADA’s system of carbohydrate exchange counting for control of diabetes.
https://www.novomedlink.com/content/dam/novonordisk/novomedlink/resources/generaldocuments/CountingCarbandMeal_EG.pdf
After getting a T2 diabetes diagnosis, my doctor gave me this method to follow. It controlled my blood glucose immediately, and I lost 50 lbs by following it for 6 months. I switched to calorie counting only after my blood glucose dropped to normal levels. Carb counting added an unneccesary layer of complexity, and effectively excluded many foods I like to eat.
I’ve been doing this for 11 years now to maintain my weight loss and smaller waistline. I only discovered Yudkin’s book a couple years ago. I was surprised to find that I had followed his CU diet for my initial weight loss.
What is the meaning of “barreling back”?
I think that caution should be used when comparing fasting insulin and post-meal levels. The insulin/blood glucose regulatory system is a bi-stable one, not an incremental release system. So we can have very distinct hormonal environments in both phases when insulin is rising, falling or holding stable. The so called paradoxical results are strong evidence that our hypothesize are in accurate or just too simplistic.
Dr Gabe Mirkin states “A study from Italy confirms that elite track and field athletes who have a family history of diabetes have larger muscles, weigh more, and are better athletes (Springerplus, 2014;3:224)” could this be a factor in this new study?
Yes. Insulin is commonly used by top bodybuilders during the hypertrophic phase of training. It s very anabolic for skeletal muscles
Hi Dr. Marcora,
Thanks for stopping by. I saw the related paper you linked on twitter and it is interesting (https://link.springer.com/article/10.1007/s00125-015-3751-0). I’ve heard that bodybuilders use insulin for muscular hypertrophy but was unsure whether it was actually effective. It does make sense because in insulin deficiency (uncontrolled type 1 diabetes) there is muscle wasting in addition to fat wasting.
“What this graph shows is that depending on the model used, one standard deviation higher post-meal insulin was associated with between about 0.01 and 0.1 standard deviation higher BMI, and this was statistically significant for most models. If I understand this correctly, roughly speaking it suggests that variation in post-meal insulin secretion explains between 1 and 10 percent of variation in BMI.”
I wonder if last sentence is correct. According to google search, standard deviation of BMI is estimated to be around 4-5 kg/m^2. Therefore, as far as I know, 0,01 and 0,1 SD increase in weight would correspond to having approximately 0,04-0,05 and 0,4-0,5 higher BMI. That is pretty low effect size. 0,01 and 0,1 SD increases in BMI (or 0,04-0,05 and 0,4-0,5 BMI increase) would therefore mean maybe 0,3 lb (0,15 kg ) and 3 lb (1,5 kg) weight gain for average person.
I am not by any means especially savvy statistically speaking, but I doubt such a small effect sizes would explain 1-10 % of variation in BMI.
Ps. Your blog is amazing. It has had huge impact on me for a couple of recent years. Understanding food reward and palatability hypothesis helped me lose around 60-65 pounds of weight pretty much permanently. Alongside that I also developed deep interest in nutrition, which consequently led me to studying clinical nutrition in European university. The Hungry Brain is definitely one of my favourite books in non-fiction genre and probably the most useful single book on nutrition I have ever read. Keep up the good word!
I don’t follow the logic of this statement:
“The total rate of release of fat from fat tissue in people with obesity is higher than in lean people, not lower (11). Neither insulin nor anything else is trapping fat inside the fat tissue of people with obesity, making it hard to understand how obesity could be caused by this mechanism.”
Most people with obesity are insulin resistant. If their fat cells are insulin resistant, then they’d release more fat from fat tissue than lean people. But that doesn’t mean they’re not releasing less fat than they would if they had no IR. They have a lot more fat to release.
BTW, I think often when you have two camps with opposing views in science, it often turns out they’re both wrong, or both right. The truth may contain elements of the insulin and the calorie hypotheses.
I mean, there’s no question the calorie hypothesis is one correct way of modeling obesity. You can’t gain weight without gaining calories so by inescapable implication you can’t gain fat without calories.
Why are people fatter than ever? People are eating more calories than ever – hard to believe that’s coincidence and of course we know it’s not coincidence.
Open questions then are:
Why do so many moderns eat so many more calories?
Is it because food is cheaper? That’s the main thing, I think.
Is it because food is more delicious? That’s the second most important reason, I think.
Is it because more of western food is made out of stuff that stimulates overfeeding through mysterious mechanisms? Possibly, but my guess is that’s an order of magnitude less important.
What’s the range of fatness variation you can find at the same level of calories for different nutrient ratios? Is there a tractable set of rules about these ratios that apply to all or most people? How much does individual variance affect this range?
The debates about these latter questions are fun and interesting, but I worry that it’s pretty distracting for ordinary people who don’t realize that calories do cause fatness, more or less. The “more or less” is unsettled, but you can use your executive function to control either more or less to achieve whatever result you desire. We have a lot of automatic behaviors, but humans are incredibly sophisticated at developing systems to override thoughtless habits.
Because of this hyper-nuanced debate, I know tons of people who literally think calories aren’t responsible for gaining weight. Once you’ve reached that stage, you’re utterly helpless, subject to the mercy of whatever fad diet latest comes across your transom.
Don’t leave out modern sedentary behavior! The Clash-of-Clans effect. Our ancestors weren’t virtual barbarians. Cordain estimates ancestral physical activity at ca 1000 kcal/day. In Fit Bit terms, 20-30,000 steps.
https://www.ncbi.nlm.nih.gov/m/pubmed/21545934/
Our digestive tracts are happiest on 3000 kcal/day. No way you can eat like that and be inactive.
I’ve been looking for mechanistic explanations for how activity triggers appetite, without success. As free fatty acids and blood glucose are stripped out of the bloodstream by metabolism they need to be replaced. But there doesn’t appear to be any direct connection between low blood sugar and appetite, and I don’t know if anyone pays attention to FFA at all.
The literature focuses heavily on bodybuilders, athletes and dieters. But it isn’t rich with material on everyone else.
minor quibble. No one denies that increasing LDL is associated with heart disease, but there are measurables that are far more associated with it. CRP and Omega3/6 for starters. so a good theory of heart disease starts with the measurables with the highest correlation. I also tend to ignore LDL because i can readily move it up or down with a 3 day dietary/exercise intervention, and so can most of those who have tried it. I think the great advantage of understanding LDL is that you can get a lower life insurance rate by testing after a 3-day fat binge.
RE: The role of insulin, has there been any discussion here about the theory of a separate “pre-prandial” or “cephalic” role for insulin? I have heard speculation that small spikes of insulin before eating (much smaller than those after eating ) can have metabolic and appetite regulating/dysregulating effects, and that the pathways/receptors/etc. are different for this role than the post-prandial one.
I believe this was the basis of the idea that 0 calorie sugar substitutes can make you just as fat as sugar, which I think is probably not the case, but wondered how much research on that idea (the general role of pre-prandial insulin) has been done.
Your posts always make me think stephan, so bear with me….
I’ve had a problem with ravenous appetite since losing weight, especially at night. It occurred to me that I had read about both lap banding and stomach stapling having an effect on reducing appetite. I thought this had to do with reducing the size of the stomach and forgot about it. However, it occurred to me that the size of the stomach is not reduced by lap banding, but simply by banding the upper part of the stomach and squeezing it. This suggested that appetite can be reduced with physical restraint of the stomach…perhaps without surgery…
It didn’t take long to find the Corset Diet and weight loss belts.
https://www.womenshealthmag.com/weight-loss/corset-diet
A lot of this is the standard weight loss diet hokum, selling overpriced garments with pseudoscientific claims. But what about the appetite suppression effect? Reading a lot of blog rolls for N=1’s showed that some people did experience appetite loss, as well as having a reduced capacity to eat. A mechanism of delayed stomach emptying is also a claim for one of the expensive belts.
So on to the practicum, which meant using materials at hand rather than buying weight lifter belts or corsets. A few years ago I picked up some old WWII surplus KD military shorts on eBay. These are high waisted, and can be cinched as tightly as you can stand just below the ribcage.
http://www.sofmilitary.co.uk/khaki-drill-kd-1941-shorts-product,7757
Other than the high waist snugness they are extremely comfortable, even to sleep in. And they do seem to delay hunger. So far so good.
Two weeks into my ghurka shorts experiment, I have no reduction in night hunger. Also no reduction – or increase – in weight or waistline. Oh well. If I persist in carrying slight calorie deficits and doing 500-1000 kcal exercise every day I guess I’m stuck with night hunger.
Not to say that there isn’t some value from wearing the tight high waisted shorts. They serve as a “waist minder”, which is especially noticeable after a large meal. I’ll keep doing it for a few months.
Hi Stephan,
Maybe Im reading it wrong, but the data on Table 2 appears to contradict (or at least influence) the author’s interpretation of the data. The highest correlation was between baseline insulin and insulin-30 (0.5194), and the highest correlation of BMI was with baseline insulin (0.4544). I don’t see any mention of this or any SNP selected that was associated with fasting insulin and tested. Shouldn’t this be very relevant for arguing that insulin-30, but not fasting insulin, is causal for obesity?
Recently read Dr Jason Fung’s book on the benefits of fasting, which I suppose is a “ketogenic diet”. His results with overweight and diabetic people is amazing- they reverse their disease states and keep the weight versus those who restrict calories and end up heavier a few years later. I did a 7 day fast, the first 2 days were really bad. There was no hunger after that and we felt amazing. We are not overweight. I thought it was interesting since it is mostly ignored in the west today